

Systemic lupus erythematosus (SLE) is a debilitating autoimmune disease that disproportionately affects women. Altered regulation of B and T cell responses and tolerance promote the development of autoreactive B cells and pathogenic autoantibodies, leading to inflammation and tissue injury, including lupus nephritis (LN). Mechanisms that promote the development of autoreactive B cells and SLE are incompletely understood. Yet this knowledge is essential for developing targeted therapies that would be preferable to currently available SLE treatment options that globally suppress the immune system, resulting in recurrent infections and increase disease-associated morbidity and mortality.
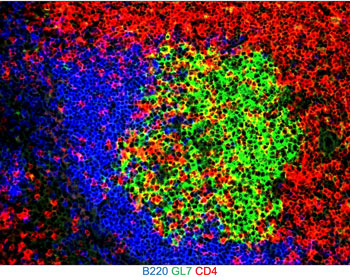
We and others previously demonstrated a B cell intrinsic requirement for TLR7 in promoting autoimmune GC and extrafollicular antibody-forming cell (EF-AFC) responses and systemic autoimmunity. Stimulation of endosomal TLRs by nucleic acids, such as TLR7 by single-stranded RNA, activates NF-κB family members, and interferon regulatory factor 5 (IRF5) and 7 (IRF7). To date, numerous studies have characterized the involvement of IRF5 and IRF5 risk variants in SLE in mice and humans. Although IRF7 is implicated in the development of SLE, IRF7-expressing cell type(s) and mechanisms by which IRF7 regulates autoreactive B cell differentiation through the EF and GC pathways, leading to autoantibody-secreting plasma cells (PCs) and autoantibody production in SLE remain unknown. IRF7 is long thought to contribute to SLE by regulating type 1 interferon (T1-IFN) production by plasmacytoid dendritic cells (pDCs), although IRF7 is also expressed in B cells and monocytes. Whether and how IRF7 may regulate autoimmune GC- and EF-derived PC responses and systemic autoimmunity through functioning in cell types other than pDCs is not known. It is also unclear whether IRF7 transcriptionally controls SLE-promoting gene programs in B cells other than T1-IFN genes. Using mouse models of SLE that promote both EF-PC and GC responses, our goal is to identify the mechanisms by which IRF7 promotes autoimmune PC and GC responses and SLE-like disease. We have developed a novel conditional and inducible mouse model on a SLE-prone background to temporally control tissue-specific deletion of IRF7 in B cells; this will help demonstrate a B cell-intrinsic requirement for IRF7 in the progression and maintenance of autoimmune GC and PC responses. Our goal in this project is to delineate T1-IFN-indpendent functions for IRF7 in regulating B cell transcription, translation, and metabolism, breaching peripheral tolerance, and promoting autoimmune GC and PC responses in SLE.