The Smeyne lab is interested in the cell biology of Parkinson’s disease, focusing on mechanisms of neurodegeneration as well as neuroprotection. Research projects examine the role of viruses in the etiology of Parkinson’s disease, the interactions of the adaptive and humoral immune systems with the innate immune system in the CNS, role of environmental toxins in experimental parkinsonism, the role of LRRK2 and alpha-synuclein as modifiers of disease progression in animal models. Our lab is also interested in mechanisms that underlie exercise-induced neuroprotection. Outside of our studies in Parkinson’s disease we are examining the effects of social isolation in adults on neuronal structure and function; as this is a growing problem facing our aging population and also has implications within our criminal justice system.

Smeyne Research

Contact
900 Walnut Street
444 JHN
Philadelphia, PA 19107
Research Projects
Effects of Viral Infections and peripheral immune system/CNS crosstalk on the Etiology and Progression of Parkinson’s Disease
The greatest influenza pandemic of the 20th century, the Spanish Flu, occurred in 1918. It is estimated that about 25-30% of the world population was infected and upward of 40 million people died in less than a year. About the same time, neurological disorders attributed to post-influenza psychosis and post-encephalitic Parkinsonism was reported both in Europe and in the USA. More than 500,000 patients who survived an influenza infection were thought to have developed some type of motor disorder; the best known of these being encephalitis lethargica or von Economo’s encephalitis. A significant number of patients who recovered from this “sleeping sickness” later developed severe parkinsonism (made famous in the book and movie “Awakenings”. Additional, there is other circumstantial evidence that viruses can act as an initiating factor in PD. IN particular, there are findings of coincident cases of parkinsonism that statistically lie outside of that expected in the general population; a phenomenon known as “parkinsonian clusters”. Other factors suggesting viral initiation is that those in professions with increased “person to person” contact have a greater than 2X risk of developing Parkinson’s disease, including doctors and nurses, teachers and members of the Clergy.

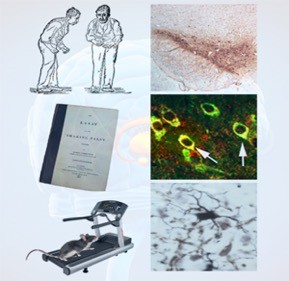
Since 2006, our lab has been examining the effects of influenza and its potential contribution to Parkinson’s disease. In collaboration with Dr. Stacey Schultz-Cherry and Robert Webster at St. Jude Children’s Research Hospital in Memphis, TN we have examined two strains of influenza, the H5N1 “bird-flu” and the H1N1 virus responsible for the 2009 pandemic flu. In regard to H5N1, we have shown that within 1 day of intranasal infection, viral particles (identified both by immunohistochemistry and electron microscopy) can be found in the lung and neurons located in the enteric nervous system. By 3 days’ post-infection (dpi), viral particles are observed in nerve fibers and their projection nuclei, suggesting that the H5N1 virus can migrate through nerve fibers to infect the CNS, without compromise of the blood-brain-barrier. Once in the CNS (starting at 3 dpi and continuing through 90 dpi), one can detect a range of parkinsonian pathologies (some transient and some permanent) that includes tremor, bradykinesia, a decrease in the number of TH-positive dopaminergic neurons in substantia nigra pars compacta (SNpc), an increase in both aggregation and phosphorylation of alpha-synuclein (the main protein in Lewy Bodies), an increase in both number and activation of microglia, as well as a time-dependent expression of inflammatory chemokines and cytokines (Fig 1).
We find a different picture with the H1N1 virus. Although this strain infects lung epithelium, we find no evidence of neurotropism. Despite the lack of virus in the brain, we did find an activation of microglia within the brain including the SNpc (Fig 2). Examination of cytokines and chemokines from blood and brain showed similar patterns of activation suggesting that the peripheral immune system was communicating with the innate immune system of the brain. This finding has opened up new insights into the etiology of Parkinson’s disease which suggests that “environmental” causes of this disease may be mediated by the peripheral immune system.
The interaction of the peripheral immune system with the innate immune system of CNS is supported by a recent study that examined the role of influenza as a Parkinson’s disease susceptibility factor. Here we infected mice with influenza and after they fully recovered administered the parkinsonian toxin MPTP. We found that mice exposed to flu had a larger loss of SNpc DA neurons after MPTP than mice that did not. More importantly, we found that if mice were immunized against influenza, the synergistic effects were ameliorated. These peripheral to nervous system interactions are being explored in relation to viruses as well as other environmental exposures, including bacterial infections.
We are now examining if influenza can also be a predisposing factor when combined with other environmental exposures including that of herbicides, insecticides and bacterial infection. We are also examining its consequences in mice carrying known parkinsonian mutations, including those in the alpha-synuclein gene and the gene encoding Leucine Rich Repeat Kinase II.
Mechanism Underlying Exercise-Induced Neuroprotection in Models of Parkinson’s Disease
There is clinical evidence that the progression of Parkinson’s disease can be slowed by physical exercise, and we have been using animal models to explore the hypothesis that such exercise can also be neuroprotective. To do so we have focused on models the induce the loss of dopaminergic neurons that underlie the motor symptoms associated with parkinsonism (tremor, bradykinesia, rigidity). Our focus on exercise derives in part from the extensive literature on the ability of exercise to increase mitochondrial respiration and antioxidant defenses, and to stimulate neuroplasticity. Using voluntary wheel running, we have shown that animals that run enough to induce neuronal hypoxia can activate biochemical pathways that induce neuroprotective proteins including activation of glycolysis, production of growth factors and induction of anti-inflammatory molecules.
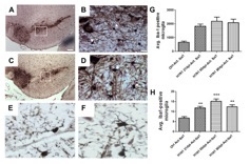
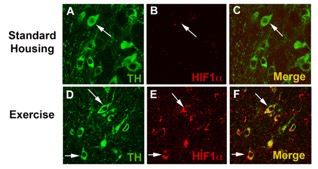
Our lab, in 2005, was the first to show (using experimental model systems) that exercise can protect SNpc DA neurons against agents that induce parkinsonism in mice. Subsequent studies performed in our lab have defined the type of exercise as well as the time and amount of exercise that is necessary to induce this neuroprotection. In 2015, we identified a mechanism underlying this neuroprotection, demonstrating the critical role for HIF1alpha, a transcription factor regulated by cellular oxygen concentration, in exercise-induced neuroprotection. HIF is a heterodimer consisting of two subunits, a constitutively present HIF1beta (Aryl Hydrocarbon Nuclear Receptor Translocator, ARNT), and HIF1a, a subunit that is expressed in hypoxic conditions. In the presence of O2, iron and ascorbate, HIF1alpha is hydroxylated by prolyl hydroxylases (PHDs) that target it for proteasomal destruction in a time as fast as 5 minutes. However, when cells become hypoxic, PHDs are inhibited and HIF1alpha is stabilized. Functionally, HIF has been shown to induce over 80 genes that function in a number of distinct biological pathways including the glycolytic pathway, erythropoiesis, angiogenesis and cell survival. We have shown that exercise induces HIF1alpha expression in SNpc DA neurons (Fig 3). To determine if HIF1alpha was critical to exercise-induced neuroprotection, we used a conditional knockout paradigm to delete this gene from postnatal neurons. We then allowed these mice 3 months of exercise (which is normally neuroprotective) and then administered the parkinsonian toxin MPTP. Unlike WT mice, HIF1alpha-CKO had a lower number of DA neuron number compared to both WT mice and also did not show any exercise-induced neuroprotection (Fig 4A). Wheel running was monitored for individual mice in exercise and exercise followed by MPTP treatment conditions for these WT and HIF1alpha-CKO cohorts. We had previously determined that 3 months of unrestricted wheel running (18k average daily revolutions) was necessary for exercise-induced neuroprotection, while 2 months of unrestricted running provided partial protection. Daily wheel running varies between and within mouse strains, so to determine whether running was protective for DA neurons, only mice running an average of 16k or greater revolutions per day for 3 months were used in this analysis. Average daily wheel revolutions (range = 16,196 to 33,766 revolutions per day) were calculated and correlated to the number of TH+ neurons in the SNpc after 3 months of exercise. We found a negative correlation between daily wheel running and TH+ cell number in HIF1alpha –CKO mice (r2=0.70, p≤0.03) in the exercise only condition (Fig 4B).
We are now examining the expression of downstream genes regulated by HIF1alpha as well as interactions with other genes in the HIF pathway, including DJ-1 and the Sirtuin family.
Role of Social Isolation on CNS Homeostasis
We are a social species; thus, it should not be surprising that the absence of social contact among those who are effectively isolated often has toxic physiological and psychological consequences. Isolation has many faces, that range from the unintended such as that which occurs as an unintended consequence of ageing and sickness (including living in a nursing home or an assisted living facility or even the isolation seen in the population of non-medical professionals who serve as caregivers) to that which is forced upon the individual (incarceration in solitary confinement). In fact, social isolation is a potent -but little understood- risk factor for morbidity and mortality, and its negative consequences are most profound among the elderly, the poor, and minorities, that are among the fastest growing elements within the U.S. population. According to a recent report from the APA, loneliness and social isolation may represent a greater public health hazard than obesity, and their impact has been growing and will continue to grow. While the psychological impact of isolation has been well described, little is known about the structural and neurochemical effects that occur in the brain following isolation. One of the big problems is that these questions are difficult to examine in human populations. For example: What changes in brain structure and function occur in individuals exposed to such conditions? How do the effects of isolation change with the duration of isolation? Does the age in which isolation occurs affect the extent of change? Are there methods by which isolated individuals can be reintroduced into a more social environment that will reduce/reverse any isolation-induced neurobiological consequences? It is for these reasons that we will make use of animal models that mimic four different conditions in which humans are grouped, including, mice housed singly in a “shoe box cage”, which we will refer to as “isolated housing” (IH, modeling total social isolation, i.e. solitary confinement), mice housed singly in a “shoe box cage” but with access to aspects of enrichment (exercise wheels and/or manipulatibles) that we refer to as “enhanced housing” (EH, modeling care in nursing facilities in which the person is oft times alone but gets access to therapy), mice housed in group conditions (4 mice) in a “shoe box cage” which we call standard housing (SH, modeling that if individuals housed in either assisted living communities or those individuals housed within a general population during incarceration) and groups of 14 mice housed in large cages (1 meter x 1 meter) with access to voluntary exercise, mazes, and toy/manipulatibles that we refer to as an “enriched environment” (EE, modeling a normal environment).
But before considering our proposal on isolation, however, it is important to ask whether observations made using animal models can inform us about any humancondition. We believe that there are several reasons to answer in the affirmative. First, rodents such as mice, like humans, are social animals preferring group over isolatedliving conditions. Second, the overwhelming majority of the human genome is nearly identical to that of the mouse. For example, of the 30,000 genes identified in C57BL/6 mice, 99% have a human equivalent. Third, the basic neuroanatomy of the mouse strongly parallels that of the human, differing mainly in the relative size of specific structures. Fourth, the key molecules that underlie neuronal plasticity are each essentially identical between the two species, including the monoamines and related enzymes, the NTFs, such as BDNF and GDNF. These similarities also exist in mechanisms for handling oxidative stress and inflammation.
Existing research indicates that there are enormous differences between rodents moved from the equivalent of a SH condition to an EE versus those maintained in “standard conditions”. Briefly, when compared to those from EE-raised, rats and mice reared in an impoverish environment show (a) deficits in learning and memory, (b) decreased neuronal complexity, (c) decrease in the number of blood vessels in brain, and (d) increase in susceptibility to several brain and behavioral conditions that emulate human diseases and psychiatric disorders, including AD and other neurodegenerative disorders, stroke, and schizophrenia. Additionally, a small but significant literature further suggests that rodent experiments are likely to be highly relevant to non-human primates. However, much of what we understand about environmental effects on the brain have been carried out in animals that were raised in an IH (or at best an SH) and then moved into an enriched environment. Our studies examining isolation better mimic the conditions of housing in humans, i.e. animals are born and raised in an EE and then later in life moved to a less enriched or completely isolated environment.
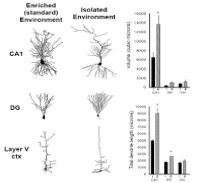
Studies in our labs support the hypothesis that housing can significantly influence brain structure and function. We examined two gross morphological features, total cell volume and total dendrite length, in neurons located in three regions of CNS from C57BL/6J mice raised in EE compared to those raised in an IH. In hippocampus, we found that pyramidal neurons from mice raised in isolation were smaller (less total volume, p≤0.01) and had reductions in their total dendrite length (p≤0.05). In the cerebral cortex, we examined layer V pyramidal cells and found similar reductions in these anatomical parameters. Conversely, we found that isolation increased the size and branching of hippocampal dentate gyrus neurons (Fig 5).
We have also examined if housing conditions can affect neurotransmitter and growth factor levels in brain, particularly in regions affected in Parkinson’s disease. We found that mice raised in the IH had lower levels of striatal dopamine as well as its metabolites DOPAC and HVA. We also have found that rats housed in an IH have reduced DA turnover and lower levels of phosphorylated TH. we also examined We examined mRNA expression of a number of growth factors, including BDNF, GDNF and IGF-1 in the basal ganglion of C57BL/6J mice raised in IH and EE. After 3 mo, mice raised in the IH had lower relative mRNA expression levels of BDNF (44% of EE mice, p<0.05, n=4), while GDNF was reduced 24% and IGF-1 increased 10% (both non-significant, n=4).