Our work focuses on two areas, the discovery and characterization of new oncogenes, and the intersection of DNA repair defects and cancer therapy.

Silver Research

Contact
Name:
Daniel Silver, MD, PhD
Position:
Professor, Department of Medical Oncology
233 South 10th Street
Office and Lab Suite 309
Philadelphia, PA 19107
Contact Number(s):
Research Projects
Screening for New Oncogenes
Multiple lines of evidence point to large gaps in our understanding of human tumorigenesis. Perhaps one of the most pressing issues is the need to identify oncogenic drivers that propel tumor development and growth when overexpressed in an unmutated state. This issue was highlighted by a landmark study of chromosomal copy number alterations present across human tumors, which documented that 51 of 76 significantly recurrent amplified chromosomal regions contain no obvious oncogenes driving the selective advantage that these amplicons must have conferred (Beroukhim et al., Nature v 463, 2010). One of our interests is to identify such wild type oncogenes and explore in mechanistic detail their contributions to human cancers.
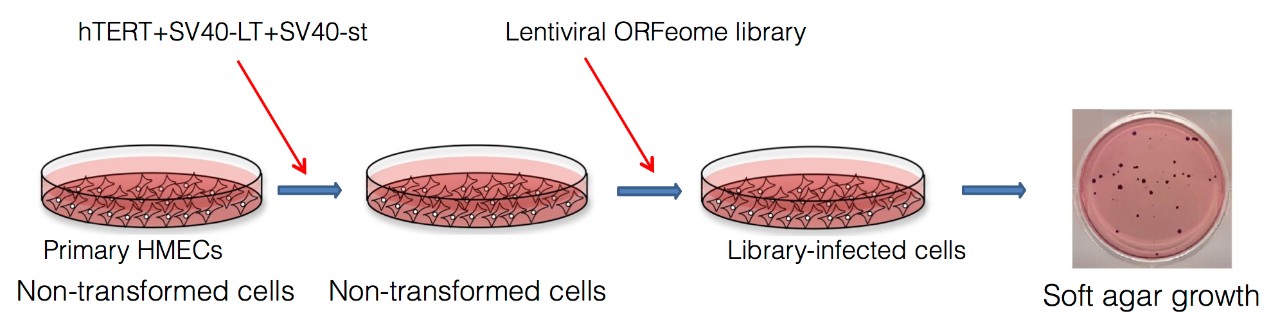
We have devised a screen to discover such oncogenes. We infect primary human epithelial cells with viruses expressing three of four genes known to be sufficient for transforming most cell types, hTERT, SV40 Large T, SV40 small t, and activated HRAS. In place of the missing gene, we infect with a pooled bar-coded lentiviral expression library constructed as an arrayed collection of 18,000 human open reading frames. We then select transformed cells from the library-infected cells by their ability to grow in anchorage independent conditions. By comparing bar code representation before and after selection for anchorage independent growth, we identify candidate oncogenes by virtue of bar code enrichment after selection. We are exploring many screen hits for their relevance to human cancers. In addition, there are many variations possible in the screen design, and we are pursuing these variations to identify additional novel oncogenes.
MECP2- Mechanism of Action & Therapy
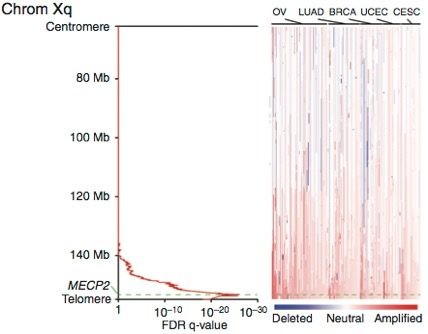
In the first application of the screen described in project 1 above, we sought genes that could substitute for activated RAS. We found a new oncogene, MECP2, which is amplified across 18% of the entire TCGA collection of human tumors (Neupane, M. et al. MECP2 Is a Frequently Amplified Oncogene with a Novel Epigenetic Mechanism That Mimics the Role of Activated RAS in Malignancy. Cancer Discovery 6, 45–58 (2016). The cancer types with the highest rate of amplification are ovarian cancer, with 38% of the cases in the TCGA showing amplification, triple-negative breast cancer with a 33% amplification rate, and lung adenocarcinoma, with a 29% amplification rate. This discovery is exciting not only because of the apparent importance of this gene in a number of malignancies, but also because we have established that its unusual epigenetic mechanism makes malignancies driven by it potentially vulnerable to an FDA-approved therapeutic, 5-azacytidine, as well as other similar drugs, and also because it suggests new rational drug combinations with 5-azacyticine.
MECP2 is one of a family of genes that is known to bind methylated CpG residues, and, more recently, it has also been shown to bind 5-hydroxymethylcytosine (5hmC). It has no previously documented role as an oncogene. MECP2 encodes two splicing isoforms that differ very slightly at their amino termini. We found that the poorly studied shorter isoform is sufficient to confer anchorage-independent growth upon human primary breast epithelial cells previously infected with viruses encoding hTERT, SV40 Large T, and SV40 small t (“N minus Ras cells”). The short isoform also activates the MAPK pathway. Strikingly, the longer, more studied isoform cannot confer anchorage independent growth upon N minus Ras cells, nor does it activate the MAPK pathway. Both MECP2 splicing isoforms are individually capable of activating the PI3K pathway. MEK inhibitors block substantially the proliferation of N minus Ras cells transformed with the short isoform of MECP2, and also prevent growth in soft agar. Importantly, when expressed together, the two isoforms drive efficient growth of N minus Ras cells as xenografts in nude mice. MECP2 binds to 5-methylcytosine, where it is thought to act as a transcriptional co-repressor, and to 5-hydroxymethylcytosine, where it may be involved in activating transcription. MECP2 DNA binding is essential for growth factor pathway activation and anchorage-independent growth. Further, a mutation in MECP2, R133C, which is known to be defective in 5-hydroxymethylcytosine binding but retains 5-methylcytosine binding, is significantly impaired in both growth factor pathway activation and its ability to confer anchorage independent growth of N minus Ras cells.
Current work is focused on understanding the mechanisms of MECP2-directed transformation and exploring the potential usefulness of therapy aimed at MECP2.
Intersection of Cancer Therapy & DNA Repair
The determinants of response to cancer chemotherapy are poorly understood, but among these determinants are the presence of defects in DNA repair in some tumors. We have a long-term interest in tumors that are deficient in homologous recombination, including tumors deficient in BRCA1 or BRCA2. We have shown that these tumors are characterized by certain recurrent chromosomal aberrations called telomeric allelic imbalance. We showed the more of these aberrations a tumor has, the more likely the tumor has a defect in homologous recombination, and the more likely such a tumor would respond to therapy targeting this defect, including PARP inhibitors and the platinum chemotherapeutic drugs. This predictive biomarker has been licensed and is now marketed as part of the Myriad “HRD” assay. Current work includes new efforts to define unique dependencies of tumors with defective DNA repair with an eye toward new therapies to exploit such dependencies.