The Naik laboratory is focused on developing therapeutic strategies to interrupt the progress of cardiovascular diseases and cancer, which are the leading causes of death in the western world. In this regard, the team has identified several novel gene products that play key regulatory roles in the progression of these diseases, with an emphasis on how these genes affect platelet functions (since platelet activity often potentiates these diseases). Using cell and molecular biological approaches, the team has characterized the potential role of calcium- and integrin-binding (CIB) protein family and junctional adhesion molecule (JAM) family members in physiological and pathological settings. Cutting edge technologies, such as the yeast two-hybrid system, siRNA, transgenic mouse models, CRISPR/Cas9, and in vivo disease models are routinely employed in the laboratory. Dr. Naik is also the Director of the Cardeza Center for Vascular Biology, Director of Integrative Physiology Graduate Program, and a member of the following Graduate Programs: Genetics, Genomics and Cancer Biology; Cell Biology and Regenerative Medicine; and Biochemistry and Molecular Pharmacology.

Naik Research

Contact
- Director, Cardeza Center for Vascular Biology
- Associate Director, Cardeza Foundation for Hematologic Research
- Professor of Medicine
Research Projects
Positive & negative regulatory mechanisms of platelet activation during thrombosis
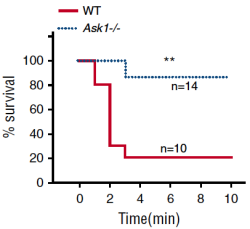
Our team has identified numerous novel regulators of platelet activity genetic ablation of which has shown protection from thrombosis (e.g. Cib1 [Naik MU, et al., 2009] and Ask1 [Naik MU, et al., 2017]) or amplification of platelet function (e.g. JAM-A [Naik MU, et al., 2012]). For example, the adjacent image from Naik MU, et al., 2017 shows how in a model of pulmonary thromboembolism, whereby clots are induced to form in the tail vein and subsequently lodge in the lungs leading to breathing cessation, Ask1 knockout mice have a much higher survival rate than their wildtype counterparts; hence deletion of Ask1 protects from thrombosis. Currently, several other genes products are being investigated through exciting in vitro, ex vivo, and in vivo methods, including carotid artery injury, cremaster injury, stroke, and Deep Vein Thrombosis models.
Regulation of new blood vessel formation (angiogenesis) by Junctional Adhesion Molecule A (JAM-A)
We have cloned and characterized a novel junctional adhesion molecule, JAM-A (Naik, U.P., et al., Biochem. J. 1995; Naik, U.P., et al., J. Cell Sci, 2001; Naik, U.P. and Eckfeld, K. J. Biol. Regul. Homeost. Agents, 2003). JAM-A is expressed in endothelial and epithelial cells and resides at the tight junctions. We were the first to demonstrate that JAM-A regulates bFGF-induced angiogenesis through its interaction with integrin alphavbeta3 (Naik, M., et. al., Blood, 2003; Naik, M. and Naik, U.P., J. Cell Sci. 2006). Using siRNA and Jam-A knockout mouse, we have shown that JAM-A is essential for bFGF-induced angiogenesis (Naik, M., et al., Arterioscler Thromb. Vasc. Biol, 2003,; Cooke, et al., Arterioscler Thromb. Vasc. Biol, 2006). We extended this work further to demonstrate that JAM-A suppresses VEGF/VEGFR2 expression on endothelial cells, thus regulating vascular permeability and angiogenesis.
Mechanism of breast and prostate cancer cell metastasis
Recently, we have found that JAM-A expression is inversely related to the metastatic ability of breast cancer cells (Naik, M., et al., Cancer Res. 2008). Over-expression of JAM-A in highly metastatic cells reduced their invasiveness; conversely, the knock-down of JAM-A in low metastatic cells increased their invasiveness. Studies are now ongoing to elucidate the molecular mechanism of this regulation. Furthermore, in collaboration with Justin Lathia of Cleveland Clinic, it has been shown that JAM-A regulates cancer stem cell function (Lathia, et al., Cell Rep, 2014).
Current Projects
- 2P20 RR015588-10
- 1R01 HL113118-02
- 1R01 HL119374-01
- AHA Grant-in-Aid
- Fraunhofer-UD Research Grant
