Uncovering the signals that regulate the decision of undifferentiated cells to embark on their differentiation pathway, and those required for their terminal differentiation, is crucial to understanding the processes of tissue development and how these mechanisms can be promoted for the regenerative repair of tissues subject to injury or disease. With the developing lens as our study model, a tissue where the precise regulation of cell differentiation and morphogenesis are key to its function, we examine these questions using molecular, biochemical and cell-imaging approaches. In our studies of regenerative repair of the lens, long-considered an immune privileged tissue, we discovered a previously unknown subpopulation of resident mesenchymal repair-modulating cells, including immune cells, and the recruitment of extrinsic immune cells in response to injury and dysgenesis. Using both a clinically relevant ex vivo mock cataract surgery model and a genetic model of lens dysmorphogenesis and opacity, we are studying the role of immune cells in the regenerative repair of the lens and how the microenvironment induces these repair cells to instead become the myofibroblasts that underlie diseases associated with fibrosis.

Menko Research

Contact
1020 Locust Street
Jefferson Alumni Hall, Room 564
Philadelphia, PA 19107

Research Projects
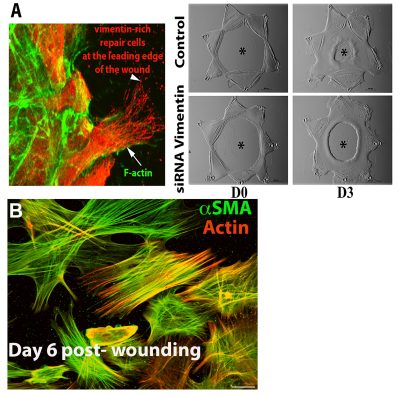
We have been investigating mechanisms of regenerative repair in studies with an ex vivo mock cataract surgery explant model developed in our laboratory. These wounded explant cultures faithfully recapitulate epithelial wound-repair, resulting in repopulation of the cell-denuded area of their endogenous basement membrane substrate. These studies led to the discovery that the lens harbors a resident population of vimentin-rich mesenchymal cells that rapidly migrate to the wound edge where they direct regenerative repair of the epithelium in a mechanism dependent on the vimentin intermediate filament protein (Image A). Cataract surgery, which this culture model replicates, often results in a fibrotic disease known as Posterior Capsule Opacification (PCO). Myofibroblasts, the principal cell type associated with fibrosis, are also induced to form in the wounded lens explant cultures (Image B). We discovered that the resident repair cell population of the lens differentiate to myofibroblasts when they encounter a microenvironment of increased rigidly. In studies with this wound-repair/fibrosis culture model we are now investigating the factors that signal the repair cells activated to direct wound-healing to alter their differentiation state and become myofibroblasts, focusing on the influence of the microenvironment and the role of their actin and vimentin cytoskeletons.
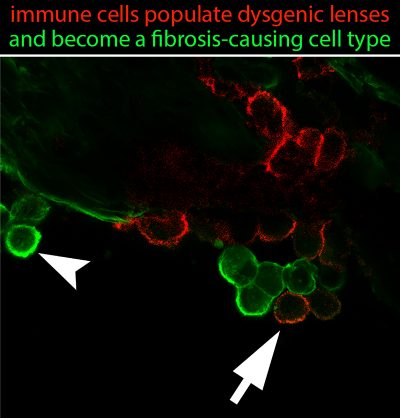
The lens, an avascular tissue, has been considered to be immune privileged, and not susceptible to the immune processes normally associated with tissue injury and wound repair. We have reexamined the concept of immune privilege in the lens in studies with our N-cadherin lens-specific conditional knockout mouse, in which loss of this cell-cell junctional protein leads to lens degeneration and fibrosis associated with development of lens opacity in the adult. The view of the lens as immune privileged was challenged by our discovery that the dysgenesis of this tissue induces an immune response, with recruitment of leukocytes, macrophages, T cells and B cells to the lens. The recruited immune cells were susceptible to be induced to differentiate to myofibroblasts, contributing to the development of lens fibrosis, as shown in the Image provided. We found that the lens is connected to the lymphatic system through the ciliary zonules that connect the lens to the ciliary body, providing a likely mechanism for immune cells to populate the lens. In addition, we observed that degeneration of the lens activates an immune response throughout the eye, including the cornea, vitreous humor, and retina; evidence that there is a coordinated protective response in the visual system. We are now investigating the mechanisms of lens immune quiescence to determine how immune cells are recruited to the lens in the absence of a vasculature.
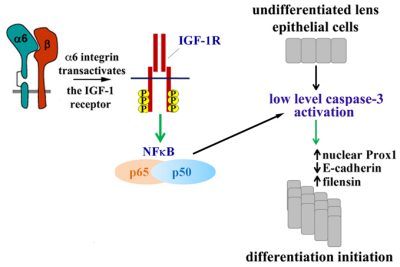
There are many different signals that must come together for a cell to carry out its differentiation program. We study a unique mechanism, discovered in our lab, required for cells to initiate their differentiation process, which is dependent on the low-level activation of caspase-3 through the canonical mitochondrial death pathway. Maintaining capsase-3 at the low-level of activation required for its function as a differentiation signal, without leading to apoptosis, involves its regulation by an α6 integrin/IGF-1R survival signaling pathway that activates of NFκB, as shown in the model provided. Low-level activation of caspase-3 induces the molecular mechanisms required for lens differentiation initiation including the nuclear translocation of Prox-1, the suppression of E-cadherin, and the expression of δ-crystallin. We are now investigating how this low-level caspase-3 signal induces targeted genetic reprogramming events that lead to the changes in gene expression required for differentiation initiation.
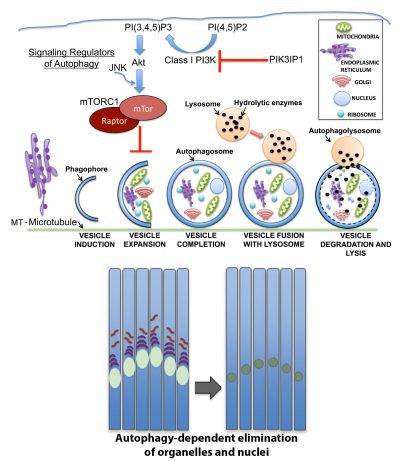
Autophagic pathways are essential to many developmental processes, yet the signals that regulate autophagy in the context of differentiation are not well understood. In the developing lens organelles are removed in a precise spatiotemporal manner from the center of the lens to create an organelle free zone (OFZ) essential to lens transparency. We discovered that the autophagic mechanisms that maintain lens homeostasis in the undifferentiated lens epithelium are distinct from those induced in maturing fiber cells for formation of the OFZ. Our studies of autophagy-linked signaling in the context of lens development showed that lens organelles are removed from the central fiber zone in a pathway that involves their disposal in autophagic vesicles following inactivation of the MAP kinase JNK, with the MTORC1 complex a downstream effector of JNK, as shown in the model provided. This MAPK is a positive regulator of MTORC1 signaling and its developmentally regulated inactivation provides an inducing signal for the coordinated autophagic removal of organelles required for lens development, and visual acuity. We are now studying the upstream regulators of this signaling pathway including PI3K/Akt, and the regulation of their activity by the novel PIK3 inhibitor PIK3IP1, for the formation of the lens OFZ.